发布时间:2022-05-31 12:00来源:www.51edu.com作者:畅畅
药监局不管你产量的大小。不过,无菌产品不一定生产全过程都无菌,如果产品特性允许,可以先在普通环境下加工,最后在洁净间内清洗、包装,再灭菌,但在普通环境下生产、包装后灭菌是不可以的,因为初始污染菌失控。如果产量小,只能尽量减小无菌控制的规模。
第三章 资源管理第七条 生产、技术和质量管理部门的负责人应当掌握医疗器械的法规、具有质量管理的实践经验,有能力对生产和质量管理中的实际问题做出正确的判断和处理。第八条 生产企业应当确定影响医疗器械质量的岗位,规定其岗位人员所必须具备的专业知识水平、工作技能、工作经验,并对从事 该岗位工作人员的能力进行评价,对未满 足规定要求的,要采取相应的措施,以满足要求。医疗器械生产操作及质量检验人员,应当经过相应技术培训,具有相关 理论知识和实际操作技能。第九条 生产企业应当具备并维护产品生产所需的生产场地、生产设备、监视和测量装置、仓储场地等基础设施以及工作环境,生产环境应当符合相关法规和技术标准的要求。第十条 若工作环境条件可能对产品质量产生不利影响,生产企业应当建立对工作环境条件要求的控制程序并形成文件或作业指导书,以监视和控制工作环境条件。第十一条 生产企业应当有整洁的生产环境。厂区的地面、路面周围环境及运输等不应对无菌医疗器械的生产造成污染。行政区、生活区和辅助区的总体布局合理,不得对生产区有不良影响。厂址应当远离有污染的空气和水质等污染源的区域。第十二条 生产企业应当确定产品生产中须避免污染、在相应级别洁净室(区)内进行生产的过程。空气洁净级别不同的洁净室(区)之间的静压差应大于 5帕,洁净室(区) 与室外大气的静压差应大于10帕,并应有指示压差的装置。相同级别洁净室间的压差梯度要合理。无菌医疗器械生产中洁净室(区)的级别 设置原则见附录。第十三条 洁净室(区)应当按照无菌医疗器械的生产工艺流程及所要求的空气洁净度级别进行合理布局。同一洁净室(区)内或相邻洁净室(区)间的生产操作不得互相交叉污染。洁净室(区)的温度和相对湿度应当与产品生产工艺要求相适应。无特殊要求时,温度应当控制在18~28℃,相对湿度控制在45%~65%。第十四条 生产厂房应当设置防尘、防止昆虫和其他动物进入的设施,门、窗及安全门应当密闭。洁净室(区)的内表面应当便于清洁,能耐受清洗和消毒。第十五条 洁净室(区)内使用的压缩空气等工艺用气均应经过净化处理。与产品使用表面直接接触的气体,其对产品的影响程度应当进行验证和控制,以适应所生产产品的要求。第十六条 生产企业应当制定洁净室(区)的卫生管理文件,按照规定对洁净室(区)进行清洁、清洗和消毒,并做好记录。所用的消毒剂或消毒方法不得对设备、工艺装备、物料和产品造成污染。消毒剂品种应当定期更换,防止产生耐药菌株。 第十七条 生产企业应当对洁净室(区)进行定期检(监)测,并对初始污染菌和微粒污染是否影响产品质量进行定期检(监)测和验证,检(监)测结果应当记录存档。第十八条 生产企业应当建立对人员健康的要求,并形成文件。建立人员健康档案,直接接触物料和产品的操作人员应当每年至少体检一次,对患有传染性和感染性疾病的人员不得从事直接接触产品的工作。第十九条 生产企业应当制定洁净和无菌工作服的管理规定。洁净工作服和无菌服不得脱落纤维和颗粒性物质,且应能有效地遮盖内衣、毛发。第二十条 生产企业应当建立对人员的清洁的要求,并形成文件。无菌医疗器械生产企业应当制定洁净室(区)工作人员卫生守则。人员进入洁净室(区) 应当按照程序进行净 化,并穿戴洁净工作服、工作帽、口罩、工作鞋。直接用手接触产品的操作人员每隔一定时间对手再进行一次消毒。第二十一条 生产企业应当确定所需要的工艺用水,当生产过程中使用工艺用水时,应当配备相应的制水设备,并有防止污染的措施,用量较大时应通过管道输送至洁净区。工艺用水应当满足产品质量的要求。第二十二条 生产企业应当制定工艺用水的管理文件,工艺用水的储罐和输送管道应当满足产品要求,并定期清洗、消毒。第四章 文件和记录第二十三条 生产企业应当建立质量管理体系并形成文件。质量管理体系形成的文件应当包括质量方针和质量目标、质量手册、本规范中所要求编制的程序文件、技术文件、作业指导书和记录,以及法规要求的其他文件。质量手册应当对生产企业的质量管理体系做出承诺和规定。第二十四条 生产企业应当编制和保持所生产的医疗器械的技术文档。包括产品规范、生产过程规范、检验和试验规范、安装和服务规范等。第二十五条 生产企业应当建立文件控制程序并形成文件,规定以下的文件控制要求:1.文件发布前应当经过评审和批准,以确保文件的适宜性和充分性,并满足本规范的要求;2.文件更新或修改时,应当按照规定对文件进行评审和批准,并能识别文件的更改和修订状态,确保在工作现场可获得适用版本的文件。3.生产企业应当确保有关医疗器械法规和其它外来文件得到识别与控制;4.生产企业应当对保留的作废文件进行标识,防止不正确使用。第二十六条 生产企业应当保存作废的技术文档,并确定其保存期限,以满足产品维修和产品质量责任追溯的需要。第二十七条 生产企业应当建立记录管理的程序并形成文件,规定记录的标识、贮存、保护、检索、保存期限、处置的要求。记录应当满足以下要求:1.记录应清晰、易于识别和检索,应防止破损和丢失;2.企业保存记录的期限应当至少相当于企业所规定的医疗器械的寿命期,但从企业放行产品的日期起不少于2年,或符合相关法规要求,并可追溯。第五章 设计和开发第二十八条 生产企业应当建立设计控制的程序并形成文件,对医疗器械的设计和开发过程实施策划和控制。第二十九条 生产企业应当确定设计和开发阶段及各阶段的评审、验证、确认和设计转换等活动。应当识别和确定各个部门设计和开发的活动和接口,明确职责和分工。第三十条 设计和开发输入应当包括预期用途规定的功能、性能和安全要求、法规要求、风险管理控制措施和其他要求。应当保持设计和开发输入记录,对设计和开发输入进行评审并得到批准。第三十一条 设计和开发输出应当满足设计输入要求,提供采购、生产和服务的依据、产品特性和接收准则。设计和开发输出应当得到批准。应当保持设计和开发输出记录。第三十二条 生产企业应当在设计和开发过程中开展设计转换活动,以使设计和开发的输出在成为最终产品规范前得以验证,确保设计和开发输出适用于生产。第三十三条 生产企业应当在设计和开发的适宜阶段安排评审,保持评审的结果及任何必要措施的记录。第三十四条 生产企业应当对设计和开发进行验证,以确保设计和开发输出满足输入的要求,并保持验证结果和任何必要措施的记录。第三十五条 生产企业应当对设计和开发进行确认,以确保产品满足规定的适用要求或已知的预期用途的要求,并保持确认结果和任何必要措施的记录。设计和开发的确认可采用临床评价和/或性能评价。进行临床试验时应当符合医疗器械临床试验法规的要求。第三十六条 生产企业应当对设计和开发的更改进行识别并保持记录。适当时,应当对设计和开发更改进行评审、验证和确认,并在实施前得到批准。当选用的材料、零件或产品功能的改变可能影响到医疗器械产品安全性、有效性时,应当评价因改动将带来的风险,采取措施最大限度避免风险,同时应当符合相关法规的要求。第三十七条 生产企业应当在包括设计和开发在内的产品实现全过程中,制定风险管理的要求并形成文件,保持相关记录。其记录应当可追溯。
流浪法师出装,英雄联盟手游小法师怎么出装
勇者斗恶龙7安卓攻略,勇者斗恶龙7安卓
lol小鱼皮肤哪个手感好,菲兹皮肤手感排行
妄想山海香料蘑菇怎么做,妄想山海臭豆腐配方和制作方法分享
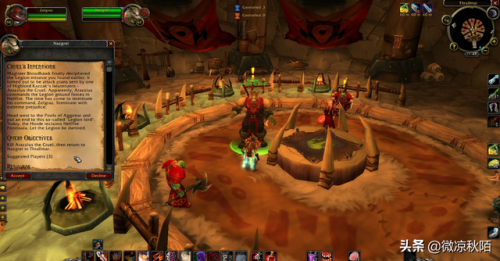
厄运之槌地图走法,魔兽厄运之槌副本入口
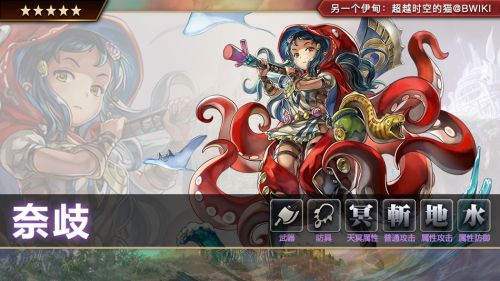
另一个伊甸奈岐角色任务,另一个伊甸奈岐技能介绍及强度测评
西部荒野稀有乌尔图斯,外域稀有精英分布图
奶茶制作方法,妄想山海烤全鱼配方和制作方法分享
洛克王国酷拉要刷多少次,洛克王国酷拉在哪
开心消消乐
类型:休闲益智
解压宝盒
类型:休闲益智
迷你世界
类型:休闲益智
恐怖奶奶
类型:休闲益智
老板挪个车2
类型:休闲益智
我的狗狗
类型:休闲益智
贪吃蛇大作战
类型:休闲益智
白块儿达人-节奏钢琴黑白块
类型:休闲益智
解压模拟大师
类型:休闲益智